Table Of Content
- 3. Docking using SILCS-MC and ML based reweighting for SAR
- 5. Membrane permeation prediction using SILCS
- Fundamental steps used in pharmacophore modeling
- My comments on ICMJE Data-sharing proposal
- Natural product-inspired strategies towards the discovery of novel bioactive molecules
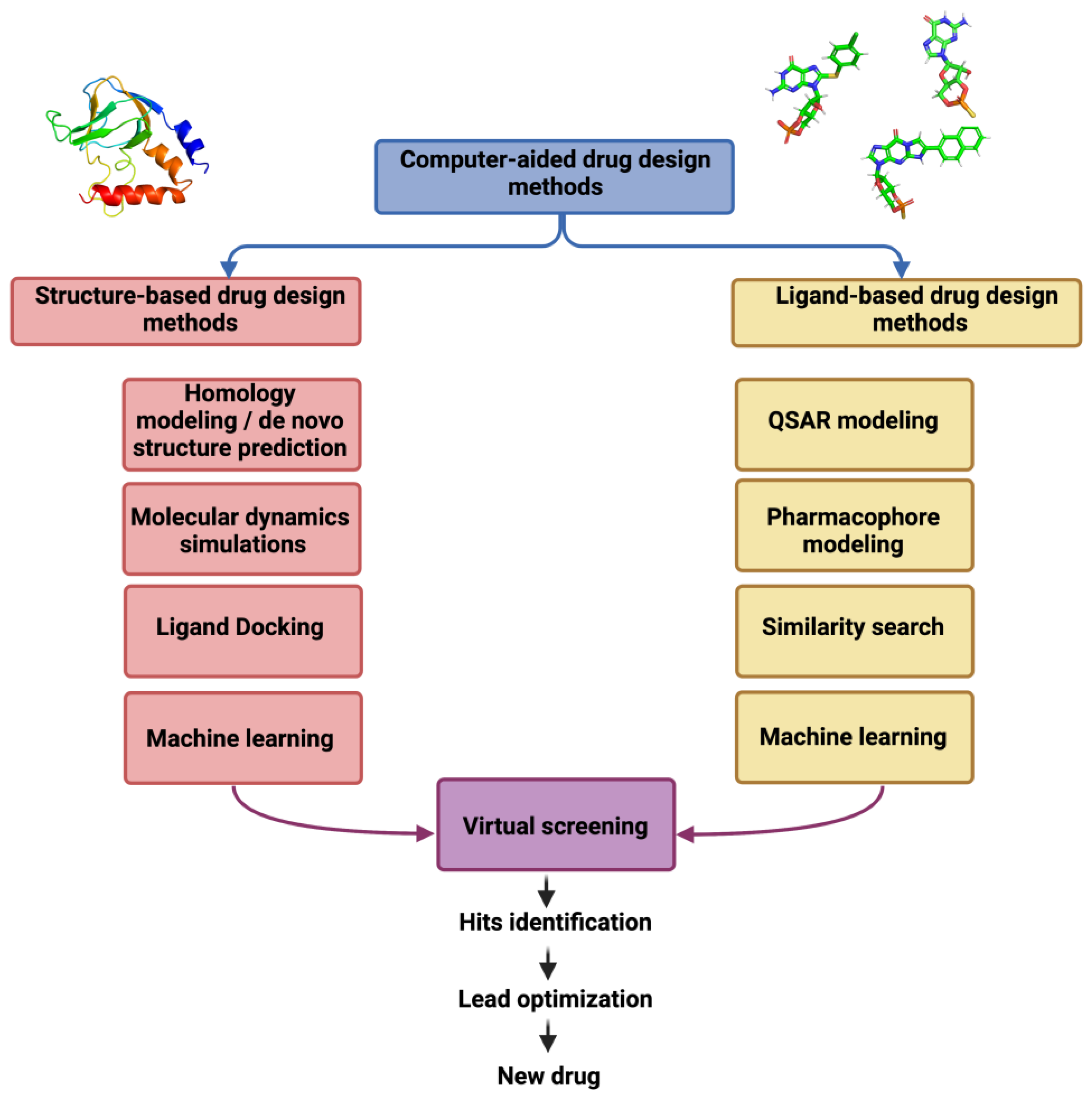
- Computer-Aided Drug Design Methods
This included studies on the mechanism behind antibiotic resistance that may help to guide the design of new antibiotic drugs to overcome such resistance. Al. studied the mechanism of a S222T mutation induced resistance of Deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) to fosmidomycin using molecular dynamics (MD) simulations (5). The MD simulations revealed the structural and energetic basis of the single mutation that induced resistance shedding light on the development of a new antibiotic compounds targeting DXR.
3. Docking using SILCS-MC and ML based reweighting for SAR
The most reliable and accurate region in the 3D structure in the protein-ligand complex is the analysis and prediction of the region in a protein where ligand can occupy its best pose. Several cavity prediction tools, such as Castro, Q-site Finder, and COACH, can provide the probable location of the binding site in a protein. The evolutionary-based approach takes the information on all related proteins and shows nearly the same binding site. Geometry-based approaches are based on the features, such as shape, hydrophobic surface, and charged surface residues, whereas an energy-based approach uses a probe to identify the favorable binding regions on a protein.
5. Membrane permeation prediction using SILCS
Biomacromolecular therapeutics, or so called biologics, need to be carefully formulated to maximize protein stability and minimize viscosity, so as to ensure both efficacy and safety for highly concentrated formulations (141). Toward maximizing stability, biologics can be formulated with excipients to help minimize aggregation and denaturation of the biologic in a solution formulation (142). To assist the rational selection of excipients for biologics, we developed the SILCS-Biologics protocol (143, 144) which combines SILCS-PPI and SILCS-Hotspots as described above to predict both PPIs that can contribute to protein aggregation and increased viscosity, and binding sites of excipients.
Fundamental steps used in pharmacophore modeling
AIDDISON™ drug discovery software Merck - Merck KGaA
AIDDISON™ drug discovery software Merck.
Posted: Tue, 05 Dec 2023 08:00:00 GMT [source]
The approach uses a pre-computed MD simulation of the hit compound-target complex from which the free energy difference due to small, single non-hydrogen atom modifications (e.g. aromatic –H to –Cl or –OH) can be rapidly evaluated (103). This is in contrast to the need for many simulations in which the chemical modification is introduced in standard FEP methods (102). SSFEP has the ability to give rapid predictions of binding affinity changes related to modifications and, thus, is quite useful for lead optimization (104). Despite the fact that numerous antibiotic drugs are available and have been routinely used for a much longer time than most other drugs, the fight between humans and the surrounding bacteria responsible for infections are ongoing and will be so for the foreseeable future.
My comments on ICMJE Data-sharing proposal
Any computational tool based on pre-defined algorithms and scripts has its limitations, and the computational tools/methods used in CADD, such as, molecular docking, virtual screening, QSAR, pharmacophore modeling, and molecular dynamics, have their own limitations [49, 85-88]. Furthermore, ADME and many toxicity prediction tools are not supported by solid experimental data, and many examples of the failure of these computational approaches can be found in the literature [89, 90]. ML algorithms are not new to the CADD area, but the increasing need for AI in areas such as image recognition and text processing promote powerful novel ML algorithms that can handle vast amount of data (29, 30). The refined graphic processing unit (GPU) architecture (31) and its growing computing power further accelerate the applications of ML, and its adaptations in CADD has erupted in recent years. ML models were trained against the resistance profiles of 14 antibiotics across three urgent pathogens using genome sequences as inputs.
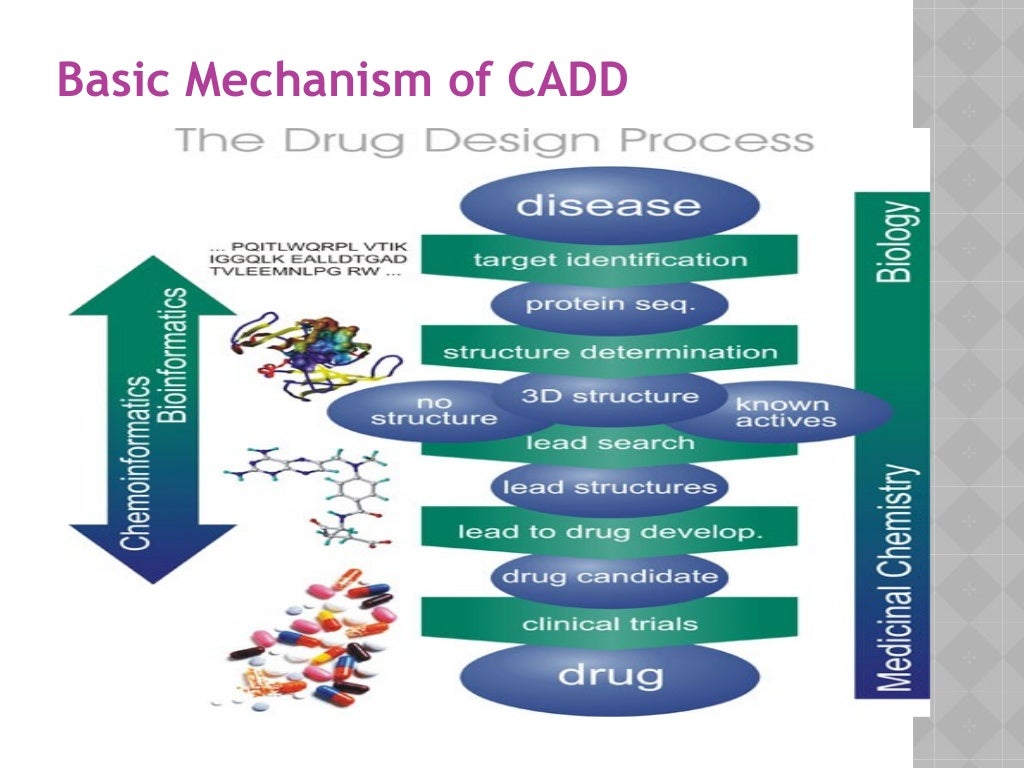
Contributing to this is the steady rise of antibiotics drug resistance leading to the need for new antibiotics (1, 2). Toward the design of new antibiotics, computer-aided drug design (CADD) can be combined with wet-lab techniques to elucidate the mechanism of drug resistance, to search for new antibiotic targets and to design novel antibiotics for both known and new targets. Notably CADD methods can produce an atomic level structure-activity relationship (SAR) used to facilitate the drug design process thereby minimizing time and costs (3, 4).
SILCS-MC then involves simply assigning the GFE value for the appropriate FragMap type to each atom in the molecule and summing those values to get the LGFE score. MC conformational sampling is then performed to allow the orientation and conformation of the ligands to relax in the field of the GFE FragMaps. This allows for SILCS-MC docking to be performed in a highly computationally efficient fashion while achieving a level of accuracy similar to highly expensive FEP methods (109).
Computer-Aided Drug Design Methods
Medicinal and toxicological investigation of some common NSAIDs; A computer-aided drug design approach - ScienceDirect.com
Medicinal and toxicological investigation of some common NSAIDs; A computer-aided drug design approach.
Posted: Thu, 29 Jun 2023 19:36:03 GMT [source]
At the final step, the top approximately 50,000 full compounds from REAL Space are docked with more elaborate and accurate docking parameters or methods, and the top-ranking candidates are filtered for novelty, diversity and variety of desired drug-like properties. Another important part of the algorithm is the evaluation of the fragment-binding pose in the target, which prioritizes those hits with minimal caps pointed into a region of the pocket where the fragment has space to grow. Like SARS and MERS, the genome of SARS-CoV-2 encodes sixteen nonstructural proteins (nsps) such as main protease (Mpro), papain-like protease, RNA-dependent RNA polymerase (RdRp), helicase etc., four structural proteins (envelope, membrane, spike, and nucleocapsid), and other accessory proteins.
Availability of the multilayer on-demand chemical space extensions (for example, supported by MADE building blocks47) can also greatly streamline the next steps in lead optimization through ‘virtual MedChem’, thus reducing extensive custom synthesis. The limited size and diversity of screening libraries have long been a bottleneck for detection of novel potent ligands and for the whole process of drug discovery. An average ‘affordable’ high-throughput screening (HTS) campaign29 uses screening libraries of about 50,000–500,000 compounds and is expected to yield only a few true hits after secondary validation.
The position and motion of every atom of the system are captured at every point in time, which is quite tough using any experimental technique. MD simulations have been extensively used in the structure-based drug discovery process as this technique helps to unravel many atomistic details such as binding, unbinding, and conformational changes in the receptor at a fine resolution which normally cannot be obtained from experimental studies [59, 60]. Further, using MD simulation it is possible to explore the dynamics of receptor-ligand interactions (association and dissociation) and quantify the thermodynamics, kinetics, and free energy landscape [61]. Virtual screening (VS) is a computational technique used for screening large datasets of molecules, and has been successfully used to complement High Throughput Screening (HTS) for drug discovery [8, 20, 21]. The major aim of VS is to enable the rapid, cost-effective evaluation of huge virtual compound databases to screen for effective leads for synthesis and further study [22]. Virtual database screening can be applied to screen large libraries of compounds using various computational approaches to identify those entities likely to bind to a molecular target of interest [23, 24].
The concept can be referred to as drug reprofiling as uncovering new indications of the either of approved/ failed/ abandoned compounds to put in use for different diseases. For larger system, more advanced MD techniques can be employed to enhance the sampling efficiency such as replica exchange methods. The protocols developed in our lab such as Hamiltonian replica exchange with biasing potentials (107) and replica exchange with concurrent solute scaling and Hamiltonian biasing in one dimension (108) are efficient replica exchange methods for use to enhance the MD efficiency.
The recently developed virtual synthon hierarchical enumeration screening (V-SYNTHES)26 technology applies fragment-based design to on-demand chemical spaces, thus avoiding the challenges of custom synthesis (Fig. 3). Starting with the catalogue of REAL Space reactions and building blocks (synthons), V-SYNTHES first prepares a minimal library of representative chemical fragments by fully enumerating synthons at one of the attachment points, capping the other position (or positions) with a methyl or phenyl group. Docking-based screening then allows selection of the top-scoring fragments (for example, the top 0.1%) that are predicted to bind well into the target pocket. This is repeated for a second position (and then third and fourth positions, if available), and the resulting focused libraries are screened at each iteration against the target pocket.
Currently, the largest commercial space, Enamine REAL Space, is an extension to the REAL database that maintains the same synthetic speed, rate and cost guarantees, covering more than 170 reactions and more than 137,000 building blocks (Box 1). Most of these reactions are two-component or three-component, but more four-component or even five-component reactions are being explored, enabling higher-order combinatorics. This space can be easily expanded to 1015 compounds based on available reactions and extended building block sets, for example, 680 million of make on demand (MADE) building blocks47, although synthesis of such compounds involves more steps and is more expensive. A large number of trials are being conducted to identify binding modes of ligands and selection of the most energetically favored poses. In order to achieve this, molecular docking tools are used to generate a set of different ligand binding poses and a scoring function is used to estimate the binding affinities of generated poses to identify the best binding mode. The energy change caused by ligand/receptor complex formation, is given by the Gibbs free energy (ΔG) and the binding constant (Kd) [49, 50].
Below, we discuss some emerging technologies and how they can best fit into the overall DDD pipeline to take full advantage of growing on-demand chemical spaces. HDAC6 is a member of the class IIb Histone deacetylases (HDACs) family and is usually found in cytosol in association with non-histone proteins [77, 78]. The implementation of CADD has been reported to result in the design of a potential inhibitor of this enzyme. In one study conducted by Goracci et al., a virtual screening approach was used to identify potential inhibitors for HDAC6, and these were then subjected to in vitro testing.
Several approaches have been developed recently to push the library size limits in HTS, including combinatorial chemistry and large-scale pooling of the compounds for parallel assays. For example, affinity-selection mass spectrometry techniques can be applied to identify binders directly in pools of thousands of compounds33 without the need for labelling. DNA-encoded libraries (DELs) and cost-effective approaches to generate and screen them have also been developed34, making it possible to work with as many as approximately 1010 compounds in a single test tube35. These methods have their own limitations; as DELs are created by tagging ligands with unique DNA sequences through a linker, DNA conjugation limits the chemistries possible for the combinatorial assembly of the library. Screening of DELs may also yield a large number of false negatives by blocking important moieties for binding and, more importantly, false positives by nonspecific binding of DNA labels, so expensive off-DNA resynthesis of hit compounds is needed for their validation. To avoid this resynthesis, it has been suggested to use ML modes trained on DEL results for each target to predict drug-like ligands from on-demand chemical spaces, as described in ref. 36.








No comments:
Post a Comment